
![[Closes 24 Nov 2107] Apply now to the OpenPlant Fund!](https://images.squarespace-cdn.com/content/v1/54a6bdb7e4b08424e69c93a1/1509564315902-TUO4I6QRWI9TT8UGSIAJ/OpenPlantTwitter_400x400+%281%29.jpg)

![[Closes 7 Mar 2017] OpenPlant Research Associate (Haseloff Lab)](https://images.squarespace-cdn.com/content/v1/54a6bdb7e4b08424e69c93a1/1486552818859-FH76MCA8SMFU93WB85RX/OpenPlantTwitter_400x400.jpg)
Applications are now open for the Biomaker Challenge 2021, which will support and fund small interdisciplinary teams of researchers to develop projects and prototypes which explore open technologies and resources focused on ‘Plants, Soil and the Environment’.
Biomaker Challenge 2020/21 Final Showcase

You are invited to join Biomaker participants and finalists for a virtual showcase of this year's projects.
Hear from the Biomaker Challenge 2020 finalists and find out more about their low-cost, open-source biological hardware projects.
Schedule:
17:00 Welcome and Introduction
17:10 Overview of 2020 projects
17:30 Finalist Presentations and Demonstrations
18:10 Q&A
18:30 Finish
Finalists
For more information please contact coordinator@synbio.cam.ac.uk
Developing a frugal and medium throughput method for assessing protein-DNA binding affinity
What are we doing?

Project collaborators Dr Susana Sauret-Gueto and Dr Eftychios Frangedakis, from the University of Cambridge.
We live in an era in which we can thoroughly investigate all the genetic material that makes up an organism, at the level of the whole genome. Understanding gene regulation, the set of processes that control the decoding of DNA, is an essential part of modern synthetic biology. Although the expression of a gene can be regulated at different levels, transcription is one of the most important steps in this complex multistage process. Transcription is the process of creating messenger sequences, known as RNA, which allow the translation machinery of the cell to build proteins according to the DNA instructions. The efficiency of transcription determines of how much of these messenger RNAs are produced.
Transcription is triggered by a collection of functional proteins known as transcription factors (TFs), which bind onto the DNA sequence in front of the part of the gene that encodes a protein (the coding sequence). This section of DNA is called the promoter. Previous research has demonstrated that the strength of a binding event between a TF and the part of a DNA sequence in the promoter that it can ‘recognise’, is pivotal in affecting the transcription of the associated gene. The strength of this binding event between a TF and its binding site (TFBS) is referred to as binding affinity. Our OpenPlant funded project, based at the Earlham Institute and the University of Cambridge, is focused of finding out how much variation in binding affinity exists in nature, and subsequently creating synthetic promoters with varying binding affinities. We aim to develop a new method to test how variation in TFBS sequence might affect binding affinity. With the knowledge gained, we can then build promoter sequences that activate transcription at the level we design, in the place in a plant we want.
Methods of assessing the binding affinity of TFs and DNA sequences already exist, but have limited scope for asking questions such as ours, either due to low throughput or high cost. One such method, which has been established for decades, is the Electrophoretic Mobility Shift Assay (EMSA). EMSA is based on visualising the travel of the bound TF-DNA complex through a jelly like matrix, or gel. Because each sequence and TF have to be made, and then individually ‘run’ through the EMSA gel, this method is difficult to scale up.

Figure 1: Comparing current protein-DNA binding assays.
Another option for testing the binding affinity of TFBS is based on a high-density DNA chip, also known as DNA microarray. This is essentially a glass slide with hundreds of thousands of short DNA sequences attached to the surface. The TF of interest can be synthesised in the lab and then hybridised with the chip. Hybridisation is just letting the binding between TF and TFBS occur as is would in the cell and, following this, the amount of TF bound on to each DNA sequence can be detected. However, the process of creating and reading such a chip is expensive and needs specific devices (Figure 1).
The aim of our work is to design and test a methodology which overcomes the shortcomings of available methods for testing TF binding affinity. Our goal is to provide a medium-throughput test of binding affinity, and we are designing our methodology to be easily replicable using affordable, readily available components. Approaching the problem from both synthetic and evolutionary backgrounds, we want to be able to test a range of binding sites for affinity, with enough replication to validate hypotheses. To do this we have developed the transcription factor relative affinity measurement pipeline (TRAMP).
Where did the ideas come from?
Inspired by the design of the DNA chip-based methods described above, we set out to design a method for assessing TF-DNA binding with as high a throughput as possible, whilst avoiding the high cost and specific requirements of the chip-based assay. We use a simple and cheap commercially available product to immobilise DNA onto the widely available lab workhorse; the 96-well plate. We have also used a recently developed method to tag our TF protein with a small peptide that, when bound to another peptide, gives off a signal. This signal is detectable in a plate reader, a piece of equipment widely used in modern labs. This new method allows us to easily quantify the amount of TF binding at a given TFBS, by measuring the brightness of the glow given off by the cumulative amount of the signal. These two components have allowed us to create a biochemical method of assaying protein-DNA binding affinity (Figure 2).
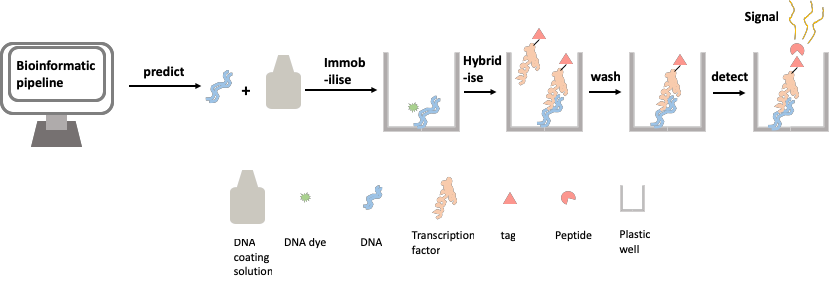
Figure 2: Assaying protein-DNA binding affinity using the transcription factor relative affinity measurement pipeline (TRAMP)
To maximise the efficiency of selecting TFBS to test in the assay we are designing, we have developed a novel computational tool. This allows us to leverage naturally occurring genetic variation in TFBS sequences gathered from publicly available population wide datasets. Instead of assaying thousands of random DNA sequences before finding a desirable one, we use several analytical methods to categorise natural variation along several parameters. We have been able to use a computational approximation of the binding event itself to score potential affinity. We also model the shape of the DNA double helix where the TFBS lies, allowing us to investigate the role of this physical property of DNA that has been suggested to be important in the efficiency of binding events.
These methods allow us to rapidly generate a set of suggested TFBS that should exhibit a range of binding affinities. This set of TFBS can be passed into the plate-based assay to be tested, and the findings used to test and further develop our understanding of binding affinity.
Where can this assay be applied?
We think that our work will be of interest to a wide range of biologists looking to understand the role of TF binding affinity in gene regulation. We hope by using TRAMP, we can find DNA sequences that exhibit different binding affinities to their corresponding TFs. Our aim is to be able to then replace the native TFBS in a promoter with the sequences we discover. We will use this approach to validate our findings, by linking the predictions and lab assays we have conducted back to transcription, the biological function we are interested in understanding.
It is our hope that our synthetic promoters will allow us to alter the activity of the promoter, meaning that when transcription occurs, the target gene will generate a different amount of messenger-RNA. If we can do this, we think our work will help scientists who want to use very specific gene expression patterns as part of synthetic biology strategies to synthesise valuable compounds like medicines. Our pipeline could also be useful for testing the effect of variation in TFBS between individuals, populations, or species.
By Dr Yaomin Cai and Dr Will Nash, Postdoctoral researchers at the Earlham Institute.
Joint OpenPlant Fund/ Biomaker call opens Monday 8 April and closes Monday 13 May
The next call for OpenPlant Fund applications is announced!
This year’s OpenPlant Fund call is joined with the Cambridge-Norwich Biomaker Challenge. More information on this joint call can be found at: https://www.biomaker.org/cambridge-norwich-challenge.
A summary of the important dates can be found below:
Call Opens: Monday 8 April
Mixer event in Cambridge: Thursday 25 April (Transport Norwich - Cambridge can be provided)
Call closes: Monday 13 May
Challenge Begins: Friday 24 May
Progress reports and presentations: Monday 29 July (OpenPlant Forum Event)
Challenge Closes/Open Technology Workshop: Saturday 2 November

An internship with the SynBio 4 Schools project
PhD student Camilla Stanton spent a three month internship, from May to August 2018, working with OpenPlant to build resources and materials for the Synthetic Biology for Schools (SynBio4Schools) project, funded through the OpenPlant Fund scheme. In this blog post she describes the project and the work that she completed during her placement.
Synthetic biology brings together researchers from a broad range of backgrounds to solve biological problems through rational design. While synthetic biology is increasingly being taught in universities, it remains under-represented in the national curriculum and teaching resources for GCSE and A-Level students. The SynBio 4 Schools project aims to solve this problem by creating a comprehensive educational resource package that teaches the principles of plant synthetic biology through practicals and case studies.

SynBio4Schools activites and write-ups on display at the OpenPlant Forum, Norwich, 2018
I got involved with the SynBio 4 Schools project through a 3-month industrial placement as part of my PhD. My role was to assess and identify what resources could be included and to begin compiling them. An obvious starting place was to explore the activities and demonstrations that researchers in Norwich and Cambridge had already developed and tested. While these resources are valuable on their own, bringing them together creates a set of interlinked resources that support one another, greatly increasing their reach and impact. It is also an exciting opportunity to get contemporary research into schools, helping inspire the next generation of biological engineers!
During my placement, I worked in collaboration with researchers to discuss ideas for how their research could be used in a teaching-style activity, whether that be an experiment, worksheet or craft-based. We also had discussions about what sort of supporting material might be useful, such as articles, interviews or case studies. It was a really enjoyable process as it gave the scientists a unique opportunity to think more creatively about their work, and I got to hear some really innovative ideas for teaching some quite complex concepts.

Some of the 3D printed virus structures from Roger Castells-Graells' OpenPant Fund Project.
I ended up focussing on writing up three activities based on work carried out by Dr Paolo Bombelli (plant microbial fuel cells), Dr Nicola Patron (genetic circuits) and Roger Castells-Graells (virus structures), which I was lucky enough to showcase at the OpenPlant Forum. This gave me the chance to receive feedback from other researchers and educators about how the materials could be made more accessible for students and provide more support for teachers and technicians. These suggestions helped shape the basic write-up template, which now includes additional investigations, sources and links to other experiments.
This was a hugely valuable experience for me - I got to explore new topics, meet people with exciting and original ideas and even got to try my hand at some design work! Although I’m now back doing my PhD, the SynBio 4 Schools project definitely doesn’t end there - we want as many people as possible to get involved.
Currently, there is a growing list of activities that cover a variety of topics from plant natural products to computational biology. But we want to showcase even more research from Norwich and Cambridge! If you have developed a resource that you would like to see included in the SynBio 4 Schools project, or you think your research could translate into an educational setting, please do get in touch! Email Colette.Matthewman@jic.ac.uk
Cell-free protein synthesis - try it with your favourite protein!
Quentin Dudley, a postdoc at the Earlham Institute, did a PhD in the Jewett lab (Northwestern University, Illinois) focused on the use of cell-free systems for the reconstitution of metabolic pathways and bioproduction of monoterpenes. Now he is using an OpenPlant Fund Award to establish cell-free platforms for protein synthesis in Norwich. Read more about this work below, and on www.biomaker.org
As part of this project he is recruiting participants for a workshop on cell-free protein synthesis to be held in mid-June in Norwich. It is an opportunity to try to express your favourite protein using a low-cost, high-throughput platform. Download the poster for details and contact quentin.dudley@earlham.ac.uk for details and questions.
Cell-free protein synthesis

Cell-free protein synthesis (CFPS) uses crude lysates of E. coli, wheat germ, and other organisms to recapitulate transcription and translation in a test tube (Carlson et al., 2012). This enables protein production at higher throughput, shorter timescales, and simpler troubleshooting compared to expression in cells. While CFPS has several pros/cons, it is particularly powerful when testing many different protein variants/mutations with an output assay that works directly in the crude cell-free reaction.
While CFPS is getting easier to implement, buying commercial kits can get expensive and troubleshooting the first time can be challenging. In response, I’m leading a project sponsored by the OpenPlant fund to establish an in-house E. coli CFPS system (~£1 / rxn) at Norwich/Cambridge and want to compare it to a commercial wheat germ kit (£12 / rxn) for expressing proteins. We are testing a range of different proteins from various plants. If you have an interesting protein you’d like to try expressing in a cell-free system, please contact quentin.dudley@earlham.ac.uk for details!)
I’ve previously worked with CFPS as a graduate student with Michael Jewett at Northwestern University. The Jewett lab is working to develop new CFPS platforms using yeast (S. cerevisiae), chloroplasts, and CHO cells. They also are improving existing E. coli-based systems to synthesize “tricky” proteins that require complex folding environments (membrane proteins, antibodies) or contain nonstandard amino acids. During my time in the lab, I used CFPS to manufacture enzyme homologs which could then be combined to prototype metabolic pathways, for example biosynthesis of monoterpenoids.
It is a very exciting time for cell-free systems. Protein yields have increased to 2 mg/mL and a commercial company (Sutro Biopharma) has reported reaction volumes at 100 L (Zawada et al., 2011). Additionally, cell-free reactions can be freeze-dried on paper and retain full activity; several groups are using this feature to develop on-demand pharmaceuticals or simple, color-changing diagnostics for diseases such as Zika virus (Pardee et al., 2016). As this cell-free technology matures, its flexibility and programmability make it an attractive opportunity for Biomaker projects and future applications will be limited only by the creativity of researchers and developers.

REFERENCES
Carlson, E. D., Gan, R., Hodgman, C. E., & Jewett, M. C. (2012). Cell-free protein synthesis: applications come of age. Biotechnology Advances, 30(5), 1185-1194.
Zawada, J. F., Yin, G., Steiner, A. R., Yang, J., Naresh, A., Roy, S. M., ... & Murray, C. J. (2011). Microscale to manufacturing scale‐up of cell‐free cytokine production—a new approach for shortening protein production development timelines. Biotechnology and Bioengineering, 108(7), 1570-1578.
Pardee, K., Green, A. A., Takahashi, M. K., Braff, D., Lambert, G., Lee, J. W., ... & Collins, J.J. (2016). Rapid, low-cost detection of Zika virus using programmable biomolecular components. Cell, 165(5), 1255-1266.
OpenPlant Fund project publishes on droplet-based microfluidics for rapid phenotyping of plant systems

Droplet-based microfluidic analysis and screening of single plant cells.
Yu Z, Boehm CR, Hibberd JM, Abell C, Haseloff J, Burgess SJ, Reyna-Llorens, I.
PLoS ONE (2018) 13(5)
PuntSeq; a toolbox and workflow to facilitate realtime monitoring of algal, bacterial and viral diversity in aquatic field work situations.
The PuntSeq team were awarded an OpenPlant Fund grant to develop a toolbox and workflow to facilitate realtime monitoring of algal, bacterial and viral diversity in aquatic field work situations. We caught up with them to find out how the project is progressing.
Full details of the project can be found on the biomaker.org website.
PuntSeq will be talking about their project at the Cambridge Pint of Science Festival. Get your tickets now to hear more about this project: https://pintofscience.co.uk/event/the-technology-behind-mainstream-headlines
Please give us a brief overview of your project (200 words max)

Water sampling from the River Cam
Year by year, Cambridge rowers, swimmers and punters obtain serious infections associated with pathogens obtained from the Cam river’s water. While an information and research framework that targets the involved microbial culprits is still lacking, our project PuntSeq is a citizen science effort that will provide an in-depth resolution of the Cam river pathogen landscape - with minimum expense!
Led by a small group of graduate students at different Life Science Departments of the University of Cambridge, we have designed a workflow for the hand-sized Oxford Nanopore MinIONTM DNA sequencing device. We are adapting software for processing large volumes of biological data from different spots of the Cam, and try to match our bacterial findings with physical measurements of the same water samples. A do-it-yourself Arduino station that combines signals from pH, temperature, turbidity and other sensors will ultimately help us understand how certain pathogens prefer to reside within particular environmental locations of the Cam.
We regularly communicate our efforts and findings through Twitter (@puntseq) and presentations at scientific conferences. Moreover, a video featuring our research ideas is also currently being produced in collaboration with Wolfson College, Cambridge.
What inspired the project?

Sampling from aboard a punt on the River Cam
Over the past years, we learned about sections of the river where people appear to often catch infections, by regularly talking to rowers and swimmers in frequent contact with the Cam. Despite the general knowledge of these unsafe areas of our river, the actual cause of the infection (i.e. the bacterial strain) remains unclear in many cases.
Up to now, taking a snapshot of the bacterial population living in a water body has required a laboratory with expensive equipment. Compared to previous sequencing machines, the Oxford Nanopore MinION dramatically reduces running expenses and is also very small, which makes it an ideal instrument for fieldwork applications. For us, this offers the opportunity to explore a new technology as well as to work interdisciplinarily by diving into a whole set of different fields from electrical engineering (Arduino measuring tool), to environmental research and the vision of personalised, data-driven health care.
How did the team meet?
Most of our members have known each other through their PhDs and previous degrees at Cambridge University. Many of us have worked together in other research projects and we share a passion for genomics research and citizen science. With an interdisciplinary combination of expertise in conservation biology, bioinformatics, engineering and physics, in situ sequencing of the Cam appeared as a really cool project for all of us to join in!
How has this project developed links between Cambridge and Norwich?
Our PuntSeq team started a collaboration with Prof. Rob Field’s laboratory at the John Innes Centre (JIC), Norwich. Amongst other environmental phenomenon, the Field lab studies algal blooms of the haptophyte Prymnesium parvum that has been associated with mass die-offs of fish in the Norfolk Broads. While the lab succeeded in associating the toxic algal blooms with infection of P. parvum by the DNA-virus PpDNAV (Wagstaff et al., 2017, Viruses), a quick monitoring system has been lacking.
Here, PuntSeq’s aim of establishing a fast metagenomics surveillance of water sources fit in perfectly. Two of our team members attended the Norfolk Broads stakeholder meeting of 2018, where we learned more about the algal blooms, exchanged our experience with DNA extraction methodology, and presented our own project of assessing the microbial community of the Cam. At this meeting, we started a collaboration with members of Rob Field’s lab to test if our approach was applicable to monitor the presence of P. parvum and PpDNAV in water in a cheap and fast manner. We hence combined our knowledge in DNA sequencing using the MinION technology, in subsequent data analysis and in engineering of environmental measurement tools to perform a metagenomics analysis on a sample of Norfolk’s Hickling Broad. As a preliminary result, we were able to draw a map of the bacterial and fungal community of the Broad, and we found a species of the toxic algae and also evidence of the virus.

The PuntSeq team joined a Norfolk Broads Stakeholder meeting, held at the John Innes Centre, Norwich
What has been your favourite aspect of the project so far?
Through our public outreach on Twitter and by regularly featuring our project at different events, we were able to discuss PuntSeq with peers and leaders in the field, for example to Prof. Nick Loman whose lab has been using the MinION to track the 2015 Ebola outbreak. We received very positive feedback and useful advice from members of the Field lab at the JIC and colleagues at the University of East Anglia (Dr Ben Wagstaff (JIC), Dr Jennifer Pratscher (UEA), Mr Elliot Brooks UEA) as well as from Alina Ham from Oxford Nanopore Technologies, which have already resulted in improvements to our DNA extraction and sequencing workflow.
Apart from this very well-received general interest in our project, we really enjoyed seeing that our first proper MinION run with the sample from the Norfolk Broads worked out - and that the results nicely confirmed our approach.
What is the biggest challenge the team have faced?
We have found it extremely challenging to extract high concentrations of DNA from river surface water, and it took us several iterations to significantly improve our low-cost protocol. Starting a MinION sequencing experiment without a laptop that fulfills the high RAM and storage requirements is very challenging and may lead to significant data loss: fortunately, Ms Lara Urban and Mr Jack Monahan from EBI have joined us and could both help with their high-performance institute machines. Since we had to do two overnight MinION runs and Lara couldn't fully dispense her computer for a full working day, the laptop-connected sequencing instrument needed to travel from our lab to her home - via Taxi! Last, waiting for >2 consecutive days of non-rain during a British spring, to only sample the Cam surface water under baseflow condition, hasn't necessarily led to a significant speed-up of our project...

PuntSeq MinION1
Is there something that came out of the project that you never expected at the beginning?
A working DNA extraction protocol, a working MinION and a working Arduino platform!
How has the OpenPlant Fund enabled the development of the project?
Through the generous funding of the OpenPlant grant, we have been able to purchase the MinION starter kit for $1000, different water DNA extraction kits, basic lab equipment and our set of Arduino sensors and wires. Moreover, Dr Colette Matthewman and Dr Jenny Molloy from OpenPlant have kindly brought us in touch with algal expert Dr. Ben Wagstaff, helping us to establish an ideal Cambridge-Norwich collaboration which will help us immensely in expanding the applicability of our approach to algal contamination of freshwater waterways. The Fund's excellent outreach network has helped us in amplifying results and messages of our project through social media channels, mainly via twitter, in addition to their kind provision of facilities for a MinION metagenome sequencing workshop that we will hold in Cambridge very soon.
How do you feel the project is progressing?
Since our PuntSeq project received its first financial funding around half a year ago, it has progressed very quickly. In these few months, our team has been able to learn about all steps that are necessary to perform metagenomics surveillance analyses, from environmental measurements over DNA extraction and MinION sequencing to bioinformatic post-processing of the data. Hereby, it is great to see how much we have learned from each other, but also entirely from scratch by reading subject literature, talking to experts and simply by trial and error. We are now at a stage where we have optimised all individual protocols to perform a major water sampling and sequencing effort at various locations of our river Cam. We expect to be able to provide a profound overview of the microbial community of the Cam by the end of Spring.
Overall, our outreach activities have been very successful although we did not present much data yet. Both scientific and non-scientific communities have shown strong interest in our project, we received a lot of positive feedback, won multiple best-poster-prizes at conferences and motivated many people to follow our progresses via Twitter (@puntseq). We are confident that this already large interest will further increase with our first results about the river Cam being released, and we are currently strengthening our public engagement efforts, e.g. by taking part in events like “A Pint of Science”, by producing a professional movie clip and conducting an online-survey on infection rates through direct contact with the Cam.
What are the future opportunities to take this project forward?
We founded PuntSeq to inform the general public about the merits of DNA sequencing, especially about the direct impact it might have on peoples' health. In future, we would ideally like to sample from multiple rivers of the greater Cambridgeshire area and beyond, producing a map of microbial communities along the length of respective waterway trajectories. We hope to share our findings with relevant environmental authorities in Cambridge and East Anglia, and to influence environmental conservation through genomics. Our team is also further streamlining the process from extraction of the aquatic DNA to sequencing with the MinION and automatic identification of potential pathogens in the field, so that non-specialists can perform these experiments and gain a deep insight into the beautiful science of microbiology.
PuntSeq team members are: Mr Maximilian Stammnitz (Department of Veterinary Medicine, University of Cambridge); Ms Meltem Gürel (Cancer Research UK Cambridge Institute); Dr Philipp Braeuninger-Weimer (Centre of Advanced Photonics and Electronics, University of Cambridge); Mr Daniel Elías Martin-Herranz (European Bioinformatics Institute); Mr Daniel Kunz (Wellcome Trust Sanger Institute); Mr Christian Schwall (Sainsbury Laboratory, University of Cambridge); Ms Lara Urban (European Bioinformatics Institute); Mr Jack Monahan (European Bioinformatics Institute); Ms Surangi Perera (Department of Physiology, Development and Neuroscience, University of Cambridge); Ms Eirini Vamva (Department of Medicine, University of Cambridge); Ms Astrid Wendler (Department of Clinical Neuroscience, University of Cambridge).
Full details of the project are at biomaker.org website. Follow the team on twitter @PuntSeq
Plant powered camera trap - are you able to take on the challenge?
With the help of funding from the OpenPlant Fund, University of Cambridge researcher Dr Paolo Bombelli together with Ms Rachael Kemp and Mr Alasdair Davies of the Zoological Society of London have launched a competition to design and manufacture a prototype of a plant powered camera trap. Deadline for proposals is 30th April 2018.

An artistic representation of a plant-microbial fuel cell
Camera trapping has been transformed by technology to become a major tool for conservationists, playing a crucial role in helping to better understand the effects of threats such as climate change and habitat loss, and supply data that can be used to inform policy and practice.
However, the current popular power sources such as battery packs and solar panels, are proving inadequate in more remote areas or in less than optimum conditions, for example in tropical forest canopies.
To overcome these challenges and further develop this area of conservation technology, this interdisciplinary team are running The Plant-Powered Camera Trap Challenge, looking to power camera traps and environmental sensors, using plant-microbial fuel cells.
Are you an architect, engineer, designer or a scientist? Are you able to design and manufacture a prototype open source plant-BES (bio electrochemical system) to power a camera trap to be used in tropical rainforests? All prototypes should be able to deliver 5v and produce 5000mC of charge per day. Submit your concepts by April 30th to receive an award of £10,000 from the Arribada Initiative and OpenPlant to build and deploy your device in the field.
If you think you can take on the challenge click here to register and find out more.
OpenPlant Fund supports project to deliver report on genetic resources in the age of the Nagoya Protocol
Dr Deborah Scott and Dr Dominic Berry of the Engineering Life project (The University of Edinburgh) have published a report "Genetic resources in the age of the Nagoya Protocol and gene/genome synthesis", based on the results of an interdisciplinary workshop held in Cambridge and involving several OpenPlant colleagues and part-funded throught the OpenPlant Fund. The workshop was dedicated to exploring emerging questions and discussions around the practice of synthesising DNA in the context of global biological diversity use and regulation, in relation to the Nagoya Protocol.

Map showing parties to the Nagoya Protocol and Biological Diversity Convention. Image by L. Tak, CC BY-SA 4.0.
Researchers in law, synthetic biology, social science and history were brought together to consider the implications of the Nagoya Protocol for Synthetic Biology and modern biotechnology. The report summarises the presentations and discussions that took place, including conversations on drivers and implications of ABS legislation, and benefit sharing and proprietary technologies.
The latter half of the report reflects on the workshop in light of the December 2016 UN Biodiversity Convention, and considers similarities and differences in the deliberations addressed at the two events.
The report ‘serves to highlight issues not yet addressed in formal negotiations and to provide additional texture to conversations already underway’.
Click to download the full report (1.4 MB PDF, 64 pages)
DIY macrophotography and embracing the challenge of video documentation

Dr Jennifer Deegan has been awarded an OpenPlant Fund grant to develop teaching materials to enable others to build duplicates of her focus stacking photography setup, and to capture images that can be used for teaching and publications in plant sciences. We caught up with her to find out what she has been up to and how her project is progressing.
Full details of her project can be found on the biomaker.org website.
Jennifer, please can you give a brief overview of your project?
Jennifer Deegan: The project follows on from my Biomaker 2017 project to build a low budget DIY Focus stacking photography system. The system takes photographs of tiny plant specimens about 2mm across, with the entire specimen in focus.

An image of a gametophyte fern, captured using the DIY Focus stacking photography system
In the past it was not possible to take photographs of such tiny specimens and have them fully in focus. This was because single images taken at high magnification had only a very shallow depth of field. With this new technique we take about 40 photographs of a tiny specimen, with the camera moving progressively towards the subject. Then all of the focused parts of the images are cut out and amalgamated together into one fully focused image.
Commercial systems are available to do this, but they are very expensive. The more affordable ones only move the camera in increments of 2 micrometres. This is not small enough for use at very high magnification. Our system is very cheap and can moved in increments down to about 1/128th of a micrometre.

The DIY Focus stacking photography system
As part of this OpenPlant project we have two goals:
- Document the construction of the focus stacking system so that others can copy it.
- Use the system to take plant photos that have never before been possible. These photos will then be made available for plant science teaching and text books.
What inspired the project?
JD: I have always been frustrated that there are no great photos of fern gametophytes anywhere. Fern gametophytes have a very interesting planar heart shaped structure that is brought about by a tightly choreographed series of cell divisions. In the literature they are usually drawn by hand, because they are too small to be photographed in full focus. During my career break to raise my son, I have been working at home as a volunteer, to try to build a system that can take good, full focus, high magnification photographs of these structures.
What has been your favourite aspect of the project so far?
JD: The judges asked me to document my system using videos rather than just in writing. This threw me for a loop initially as I have never made video and didn't have the equipment. However, I have managed to cobble a system together, and am loving my new craft. The time, nuance and attention to detail that is needed to make a short video is amazing. The photo below shows the many photo, video and sound files that I had to record and line up in order to create one short video. I'm now the proud owner of a YouTube channel. (You can visit it, and the other documentation on GitHub and Hackster via www.chlorophyllosophy.uk)

Editing videos that explain how the focus stacking system works
What are the biggest challenges you have come across?
JD: There have been a lot of challenges, particularly with the transition from written documentation to video.
The biggest problem is that my laptop is ten years old and is a bit slow for editing video. It cannot play my videos at full speed, so I have to upload them to YouTube between editing session to see what they look like. Saving the files out for upload to YouTube takes 2.5 hours for each video, so it is a slow process.

The DSLR filming the focus stacking setup, with decoy camera body in place
One of my funniest solved problems is that my DSLR is the only camera that I have that can record video, but it also has to appear in the videos. I got around this problem by putting my 27-year-old film SLR as a body double in the videos. The photo to the right shows my DSLR filming the focus stacking setup, with decoy camera body in place. It’s great fun editing the sound of the camera shutter into the finished video.
My other challenge is making these rather technical videos engaging to watch. There is a definite risk of them coming over as a bit dry, and so I try to keep them short and make the images interesting. I think that if I can improve my editing equipment at some point, I could make my videos much more engaging.
I’m really enjoying making educational videos and would like to keep doing this work after the end of the OpenPlant grant. I’ve been in touch with the University Public Engagement Office, who have been very helpful, and I’m hoping to learn some tips from them.
You have been awarded both a Biomaker Challenge and OpenPlant Fund grant. How have these enabled the development of the project?
JD: My work absolutely could not have been done without these grants. Most of the work has been done through collaboration, volunteer labour, and home engineering. However, the grants paid for the microscope objectives. Without these amazing lenses, I could not have done the work.
How do you feel the project is progressing?
JD: I think it's going very well. I have four good videos already online, and a lot of written documentation. I have registered a new domain (www.chlorophyllosophy.uk) as a central doorway to all of the material, and I still have lots of ideas for other videos to make.
Two out of three of my lenses have arrived and I am looking forward to taking some great photos. My Utricularia gibba (bladderwort) plants are growing well in their casserole dish. Utricularia gibba is a small, carnivorous aquatic plant that develops traps to capture its prey. They are being studied by my collaborator Christopher Whitewoods at the John Innes Centre and I have already taken my first few photos of them, as the new traps develop. The traps have a beautiful structure, and as an aquatic plant, will be a great challenge to photograph.
I hope soon also to visit the Sainsbury Laboratory in Cambridge to photograph the trichome mutant phenotypes in Arabidopsis thaliana, belonging to my collaborator Aleksandr Gavrin. I really look forward to the challenge of photographing trichomes, that will have other trichomes behind to confuse my software.
I have also just sewn a new batch of fern spores and those plants will be a real treat to photograph when the time comes.
What are the future opportunities to take this project forward?
JD: One of the biggest pitfalls for photographers is that they become so fascinated by the stream of newer and better camera equipment, that they forget to actually take any photos. I think that in the next couple of years, it's very important that I actually take the time to take some photographs. With this new technology that I have built, and with the opportunity of my volunteer labour, these will add hugely to the body of research knowledge.
Jennifer's project is also documented on Github: https://github.com/BioMakers/Gametophyte-Fern-photography-2018/blob/master/README.md
Eleven projects pitch for funding from the OpenPlant Fund

Aleksandr Gavrin pitching his proposal.
Friday 1 December 2017, Norwich, was the day of the pitches for the 5th round of OpenPlant Fund proposals – and what an exciting set of proposals they were. Eleven proposals were pitched, ranging from development of plant tools and methods, to cell-free protein production, software and hardware development, training, and development of resources for schools in Ghana.
The OpenPlant Fund is rapidly building a dynamic community of early career plant synthetic biologists. The Fund has awarded over 60 micro-grants between 2015 and 2017 to projects facilitating exchange between University of Cambridge, the John Innes Institute and Earlham Institute in Norwich and a range of external collaborators for the development of open technologies and responsible innovation in the context of synthetic biology. Through these awards, OpenPlant aims to promote plant synthetic biology as an interdisciplinary field. This latest round of “high quality, innovative and novel ideas” – as judge Richard Hammond of Cambridge Consultants put it – highlights the engagement, motivation and drive the is present within the local community. More information on the Fund can be found at www.openplant.org/fund and documentation of OpenPlant Fund projects can be found at www.biomaker.org.

Fern gametophyte photographed by Dr Jennifer Deegan using her focus stacking photography platform. More information, images and project documentation can be found through http://chlorophyllosophy.uk/
Tools for plant synthetic biology
The first talk, coming to us via skype, pitched for funding to further develop a focus stacking photography platform for teaching and publication in plant sciences. Impressive images of fern gametophytes showed the current scope of the platform developed through the Biomaker Challenge. Presenter Jennifer Deegan (University of Cambridge) made full use of skype by demonstrating the hardware setup, explaining how it would be further developed to expand its scope, and how it would be adapted to build a cheap system for schools.
Next up, Aleksandr Gavrin (Sainsbury Laboratory, University of Cambridge) presented a proposal to make stable transgenic Medicago truncatula lines in which actin is tagged with a reporter gene as a tool for legume researchers. In another legume-focused project, Abhimanyu Sarkar (John Innes Centre) proposed to establish a transformation system for the orphan crop Grass-pea. While there were some challenging legal questions surrounding the shareability of the system, the judges recognised the urgent need for new developments in transformation.

Image by Pablo Ramdohr, shared under licence CC BY 2.0
Cell-free biology
Proposing to compare cell-free and plant expression systems for protein expression, Susan Duncan (Earlham Institute) pitched a project that would analyse synthesis of proteins, focussing specifically on transcription factors. New collaborations between groups in Norwich and Cambridge will provide Susan with a variety of transcription factors to test.
In a related, but “very independent” project, Quentin Dudley (Earlham Institute) proposed to compare protein synthesis in two different cell-free systems, E.coli and wheat germ lysates. The project aims to gather data on yield vs cost of the two systems. He extended on open invitation for people to ask him “can you try my protein”. So, get in touch if you’d like your plant protein to be tested in Quentin’s cell-free systems.
The third cell-free proposal came in via skype, with Clayton Rabideau (University of Cambridge) rubbing the sleep from his eyes to pitch from the US in the early morning hours. Clayton pitched for funding to develop a hardware system called Open-Cell, using machine learning together with microfluidics-based cell-free screening assay technology for screening of enzyme activity.
Computation and training
A third theme that came out through the pitches, was the need for computation, software development and training. Chris Penfold (University of Cambridge), who had arrived straight off a plane from Venice, proposed an ambitious project to develop a suite of computational tools to simulate large gene regulatory networks in plants and mammals. These tools aim to improve rational design and predictability in synthetic biology.

Jan Sklenar (The Sainsbury Laboratory, Norwich) presented a proposal to bring together proteomics experts and bioinformaticists with expertise in R software. To do this, the group propose a series of workshops for knowledge exchange and training to help both disciplines understand each other. Following these workshops, the team will work together to integrate the ‘R for Proteomics’ package, developed at the University of Cambridge, into Norwich proteomics workflows and further develop the software suite. Jan’s driving motivation for the project is to “be more efficient” and require “less manual interference” for proteomics analysis.
A final computational project was pitched by Aaron Bostrom (Earlham Institute) who talked about mutant worms and Raspberry Pi’s in a proposal to develop a training programme designed around sensing hardware for data collection and machine learning for plant synthetic biology projects.

An artistic representation of a plant-microbial fuel cell, submitted in Paolo Bombelli's proposal
International activities
Two energetic presenters pitched projects focussed on engaging directly with an international group. Paolo “the plant electrician” Bombelli (University of Cambridge) pitched for match-funding to enable him to run an international biodesign competition for the development of prototypes for a plant-microbial fuel cell to be used in remote jungle regions as an environmentally friendly power supply for a sensor and camera-trap to be used by Zoologists.
Waving his hands as he introduced himself, PhD student Hans Pfalgraz (University of East Anglia and John Innes Centre) proposed a project, working with Kumasi Hive innovation hub and the Lab_13 Ghana practical science education project, to take inspiration from previous OpenPlant projects and develop open source practical teaching activities, testing these in Ghana and then making more widely available for schools in other low-resource settings.
What the judges say
“This was a great event and I thoroughly enjoyed it. It felt like we visited all four corners of science in a couple of hours. The proposals were of a high standard and well presented with some fascinating new ideas to understand and discuss. Well done to all involved.’”
“It was a great day, very good science, creativity and a warm welcome. Thanks for the invite!”
“We heard a number of compelling and original ideas, the majority being led by early career researchers. It was particularly impressive to see so many new collaborations and networks being built, both between the Open Plant Research Institutes and with external partners.”
[Closes 24 Nov 2107] Apply now to the OpenPlant Fund!

The OpenPlant Fund is now open to proposals for innovative, open and interdisciplinary projects relevant to plant or in vitro Synthetic Biology. Projects run for six months and can include biological research, hardware prototyping, software, outreach, policy work and training.
For this round applications focused on training and knowledge exchange are especially encouraged.
The deadline is 24 Nov 2017 for projects led from University of Cambridge or Norwich Research Park with external collaborators welcome.
Download: Poster | Info Sheet
Each successful project will receive up to £5k, with £4k up front and an additional £1k for follow-on and outreach after reporting. PhD students and postdocs are particularly encouraged to apply.
A wealth of tools, technologies and methodologies have been developed for plant and cell free Synthetic Biology, including those developed through OpenPlant, the OpenPlant Fund, the Biomaker Challenge and complementary efforts. In the current OpenPlant Fund call, we are encouraging applications for projects that will provide training or knowledge exchange to broaden the use of plant and cell-free synthetic biology tools, techniques and technologies. Information about previous OpenPlant Fund projects are available on www.biomaker.org.
For more information see https://www.openplant.org/fund/ and join the upcoming mixer event on Thursday 9 Nov 2017. If you are interested in submitting a proposal or have any questions, please email colette.matthewman@jic.ac.uk.
Want to learn more and find collaborators?
OpenPlant Fund mixer and a light-hearted look at training!
4pm on Thursday 9 November, The Rec Centre Bar, John Innes Centre, Norwich
RSVP to this event here >>
This event provides an introduction to the opportunities and a chance to present your initial proposals and to meet potential collaborators over drinks and pizza. We will also have a light-hearted look at training, including different models and effective communication of technical details. Come along to learn some tips and tricks in this fun and informative training session, and to network and meet potential collaborators. More details to follow.
Cafe Synthetique: Towards engineering circadian rhythms
6pm on Monday 20 November, The Panton Arms, 43 Panton Street, Cambridge
RSVP to this event here >>
Help will be on hand to answer any questions you might have as the deadline for applications approaches and to find last minute partners for your teams!
Eligibility
Applicants should be graduate students or postdoctoral workers at the University of Cambridge, the John Innes Centre or The Sainsbury Laboratory. The team must be interdisciplinary, must contain members from both Norwich and Cambridge and may contain external collaborators of any type. Applicants must have agreement from their research supervisor and cost-code sponsor that the proposed project and management of the allocated funding will fit with their existing work. All proposals must lead to tangible, publicly documented and open outcomes, which could include (but are not limited to) the following:
- Design files and prototype for a hardware project
- Software development and documentation
- White paper arising from a workshop
- Educational resource
- Synthesis and sharing of useful DNA parts or vectors.
For more information and to apply see the OpenPlant Fund webpage
Low cost and open source multi-fluorescence imaging system for teaching and research in biology and bioengineering
Former OpenPlant Fellow Dr Fernan Federici, former OpenPlant PDRA Dr Tim Rudge and colleagues have recently published a pre-print for their low cost and open source multi-fluorescence imaging system for teaching and research in biology and bioengineering, supported by the OpenPlant Fund.
Nuñez, Isaac, Tamara Matute, Roberto Herrera, Juan Keymer, Tim Marzullo, Tim Rudge, and Fernan Federici. "Low cost and open source multi-fluorescence imaging system for teaching and research in biology and bioengineering." bioRxiv (2017): 194324

Examples of images of bacterial colonies and cell-free systems using the microscope. Credit: Federici Lab
Abstract
The advent of easy-to-use open source microcontrollers, off-the-shelf electronics and customizable manufacturing technologies has facilitated the development of inexpensive scientific devices and laboratory equipment. In this study, we describe an imaging system that integrates low-cost and open-source hardware, software and genetic resources. The multi-fluorescence imaging system consists of readily available 470 nm LEDs, a Raspberry Pi camera and a set of filters made with low cost acrylics. This device allows imaging in scales ranging from single colonies to entire plates.
We developed a set of genetic components (e.g. promoters, coding sequences, terminators) and vectors following the standard framework of Golden Gate, which allowed the fabrication of genetic constructs in a combinatorial, low cost and robust manner. In order to provide simultaneous imaging of multiple wavelength signals, we screened a series of long stokes shift fluorescent proteins that could be combined with cyan/green fluorescent proteins. We found CyOFP1, mBeRFP and sfGFP to be the most compatible set for 3-channel fluorescent imaging. We developed open source Python code to operate the hardware to run time-lapse experiments with automated control of illumination and camera and a Python module to analyze data and extract meaningful biological information.
To demonstrate the potential application of this integral system, we tested its performance on a diverse range of imaging assays often used in disciplines such as microbial ecology, microbiology and synthetic biology. We also assessed its potential for STEM teaching in a high school environment, using it to teach biology, hardware design, optics, and programming. Together, these results demonstrate the successful integration of open source hardware, software, genetic resources and customizable manufacturing to obtain a powerful, low cost and robust system for STEM education, scientific research and bioengineering. All the resources developed here are available under open source license
My OpenPlant Experience: Outreach, Engagement and 3D printing
Guest blog post by Roger Castells-Graells about his OpenPlant Fund project “Accessible 3D Models of Molecules”. Roger recently won a UEA Engagement Award in recognition of the work he has done both with OpenPlant and beyond.

PhD student Roger Castells-Graells in the lab
My name is Roger and I am a PhD student in Prof. George Lomonossoff’s lab at the John Innes Centre in Norwich. My research project is about the production of virus-like particles to understand viral dynamics for future applications and to generate new bionanotechnological tools. I have a passion for science communication and public engagement and I have had numerous opportunities to communicate my science in Norwich, the UK and abroad since the start of my PhD.
My OpenPlant experience started in September 2016, when I attended a great Co-Lab workshop organized by the Open Science School and funded by an OpenPlant Fund. With this opportunity I had the chance to interact with scientists from different fields and also with designers and artists. It was an enriching experience and we developed a project called VRICKS (Virus Bricks) that aimed to generate tools to explain viruses in educational ways, like for example with paper models.
Following up from this workshop, in October 2016, I organized an activity for the Norwich Science Festival, together with Jenni Rant (The SAW Trust) and Colette Matthewman (OpenPlant), where we recreated the assembly of proteins into a virus protein coat using materials like paper and plastic, which represented the subunits of the virus. The public contributed to the assembly of a virus model, they learnt about related research from the Lomonossoff lab and they took home a build-at-home model. Over one hundred people participated in the activity during the weekend, making it a roaring success.

Presenting the virus activity and engaging with people at the Norwich Science Festival
Following up with the interest to build tools to explain biological processes, such as virus assembly, I decided to apply for and OpenPlant Fund with the project “Accessible 3D Models of Molecules”. The project team is a multidisciplinary team (molecular biology, bioinformatics and engineering) of students from JIC and University of Cambridge and with this fund we are developing models of viruses and proteins using 3D printing technologies.

3D printed virus models for the OpenPlant Fund project
Recently I presented some of the virus models in a high school with students aged 12 to 16 years old. The students enjoyed being able to handle and compare representations of real virus structures and were amazed that some of these structures were only discovered this year. When the school teacher was asked about how the use of educational 3D models in the classroom could benefit the learning process he answered that first of all it creates excitement and focuses the attention of the students. It is something completely new! It contributes to the understanding of three-dimensional models and gives the students a better sense of the reality of the object. Furthermore, it allows the students to calculate scale as it is possible to touch, measure and compare different models.
I was invited to speak at the Pint of Science Festival in Norwich in May, and gave a talk entitled “20000 Leagues under the microscope: Viruses & Nanomachines”. At the event, I passed around several models of 3D printed viruses and the public loved having the opportunity to handle them. It was a great experience and we received really positive feedback. I want to thank the organizers of Pint of Science for such a great event!
As a result of all of these activities, I was recently awarded a UEA Engagement Award 2016/17 for contribution to Public & Community Engagement, which I am very proud of.

Norwich Pint of Science Festival tweets

With thanks to my supervisor Prof. George Lomonossoff, OpenPlant and all the people that have helped, encouraged me and opened up opportunities in this last year.
[Closes 30 June 2017] Apply now for the OpenPlant Fund
The OpenPlant Fund is now open to proposals for innovative, open and interdisciplinary projects relevant to plant or in vitro Synthetic Biology. Projects run for six months and can include biological research, hardware prototyping, software, outreach and policy work.
Each project will receive up to £5k, with up to £4k up front and an additional £1k for follow-on and outreach after reporting. PhD students and postdocs are particularly encouraged to apply and external collaborators are welcome.
The aim of the fund is to promote the development of plant Synthetic Biology as an interdisciplinary field and to facilitate exchange between the University of Cambridge, the John Innes Centre, and the Earlham Institute for the development of open technologies and responsible innovation in the context of Synthetic Biology.
More information can be found here: https://www.openplant.org/openplant-fund/
OpenPlant Fund opens to applications for £5000 grants on plant or cell-free synthetic biology

OpenPlant Fund offers £5000 to support open, interdisciplinary and innovative projects to engineer plant biology. Applications are now open until 1 Dec 2016 for projects led from University of Cambridge or Norwich Research Park with external collaborators welcome. For this round applications focused on cell-free synthetic biology are also encouraged.
The aim of the OpenPlant fund is to promote the development of plant Synthetic Biology as an interdisciplinary field and to develop open technologies and responsible innovation in the context of plant Synthetic Biology.
This call is also encouraging applications related to use of cell-free extracts from bacteria, plants, yeast or other organisms to transcribe and translate engineered DNA. Cell-free synthetic biology is gaining popularity for prototyping genetic circuits and metabolic pathways and has many applications from production of biologics to paper-based diagnostic tests and biosensors.
OpenPlant Fund teams facilitate exchange between The University of Cambridge, the John Innes Centre, The Earlham Institute and The Sainsbury Laboratory and therefore are led by researchers from these institutions, but are open to all external collaborators.
Download: Poster | Flyer | 2015/16 Report
Apply now >>>
Want to learn more and find collaborators?
Join us at a mixer event at the Panton Arms on 21 November 2016, 18:00-20:00. Great talks from a previous funded project on microfluidics and from the Cambridge spin-off Sphere Fluidics plus an opportunity to pitch your idea or find a team to join!
Eligibility
Applicants should be graduate students or postdoctoral workers at the University of Cambridge, the John Innes Centre or The Sainsbury Laboratory. The team must be interdisciplinary, must contain members from both Norwich and Cambridge and may contain external collaborators of any type. Applicants must have agreement from their research supervisor and cost-code sponsor that the proposed project and management of the allocated funding will fit with their existing work. All proposals must lead to tangible, publicly documented and open outcomes, which could include (but are not limited to) the following:
- Design files and prototype for a hardware project
- Software development and documentation
- White paper arising from a workshop
- Educational resource
- Synthesis and sharing of useful DNA parts or vectors.
For more information and to apply see the OpenPlant Fund webpage.
Guest Post: If you can protoplast, you can encapsulate
The following is a guest post from the SynBio Fund project ‘Development of a microfluidic device for high-throughput analysis of genetic circuits in plant protoplasts’ from by Steven Burgess, Ivan Reyna-Llorens, Christian R. Boehm, Sara Abalde-Cela and Paul Bennett. A continuation project, entitled ‘Establishing 3D Printed Microfluidics for Molecular Biology Workflows’, was funded through the OpenPlant Fund in 2016 and recently a second OpenPlant Fund grant was awarded for the project ‘Plant-ProChip 2.0: High throughput transformation of plant protoplast’.
You can read the original post on their project blog here >>
We started this project with the aim of testing whether it is possible to use microfluidics to analyse plant protoplasts, and I think we now have the answer. After numerous rounds of testing we have improved our working method and are now able to routinely isolate and encapsulate protoplasts. This has been done for two model plant species including A. thaliana, and everyone’s favorite Bryophyte –Marchantia polymorphia, the workhorse of the OpenPlant Project for plant synthetic biology (Figure 1).
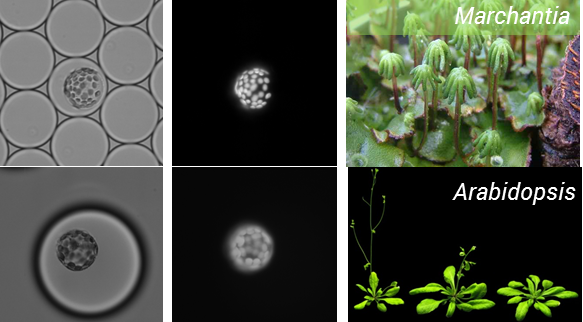
Figure 1: Encapsulation of protoplasts from model plant species
So the take home message from this project is – if you can protoplast you can encapsulate! But the story does not end here. To be of real use, this process needs to be coupled to transformation of protoplasts. As a result, we teamed up with Oleg Raitskinfrom Nicola Patron’s group at the Earlham Institute. Oleg has been optimizing protoplast isolation and transformation using Nicotiana benthamiana and had a couple of tips for improving isolation, including the use of a cork borer instead of scalpel blade for cutting up tissues to minimize mechanical damage, cutting tissue when submerged in the enzyme mix and using a high ratio of DNA to protoplasts during PEG transformation.
This was a fruitful collaboration, Oleg managed to transform protoplasts with a nuclear targeted Venus reporter and these were encapsulated by Ziyi in the Chemistry department (Figure 2).
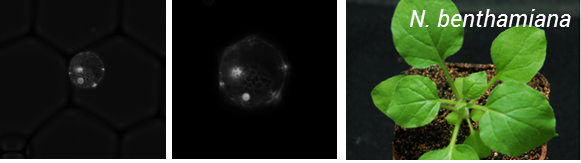
Figure 2: Encapsulation of N. benthamiana protoplats expressing a nuclear targeted Venus reporter
So putting all this work together, we have in hand a simple, but very powerful system that opens up a whole range of possibilities for rapid phenotyping in plants (Figure 3).

Figure 3: Schematic of microfluidic analysis of plant protoplasts and some of the potential applications.
One of the stipulations of the project was to pursue science in an open manner, so we have been putting up information on the website protocols.io. I highly recommend checking out the site if you haven’t done so already, it has a great set up for disseminating protocols. Further we believe microfluidics is a great technique, so would encourage others to have a go as well!
Looking to the future there are still a few things we would like to work on, The project was briefly presented at Cambridge’s Cafe Synthetique meet-up and we had some great feedback, such as trying Calcium alginate encapsulation as a means of improving protoplast viability. Sorting of protoplasts is the next major goal, and requires redesign of a new chip, and finally improving the efficiency of protoplast transformation by developing an on-chip procedure would be a big advantage. This round of our project has come to an end, but stay tuned for future developments.
Finally I want to finish this piece with a big thanks to Cambridge Synthetic Biology SRI for funding the work, it has been a great experience, and to encourage anyone who is interested in protoplasts or microfluidics to get in contact, we are always happy to chat!
OpenPlant Plant Fund 2016 - Pitches on Mon 14 March announced!
 We've got a fantastic line-up of teams pitching for the OpenPlant Fund - £5000 grants to support innovative, open and interdisciplinary projects to engineer plant biology. All are welcome to hear the teams 13:00-16:30 on Mon 14 March in the Large Lecture Theatre, Department of Plant Sciences, University of Cambridge. We'll be sticking strictly to time, so feel free to drop in even if you can only make it for a couple of pitches.
We've got a fantastic line-up of teams pitching for the OpenPlant Fund - £5000 grants to support innovative, open and interdisciplinary projects to engineer plant biology. All are welcome to hear the teams 13:00-16:30 on Mon 14 March in the Large Lecture Theatre, Department of Plant Sciences, University of Cambridge. We'll be sticking strictly to time, so feel free to drop in even if you can only make it for a couple of pitches.
Pitch Timetable
14 March 2016, 13:00 - 16:30 Large Lecture Theatre, Department of Plant Sciences, University of Cambridge
| Time | Project Title |
| 13:00 | Opening remarks |
| 13:15 | Hot Tomato: Complementation of the Capsaicin Biosynthetic Pathway to Engineer Spicy Tomatoes |
| 13:30 | Synthetic Biology for Schools: A multidisciplinary approach |
| 13:40 | Implementation of a synthetic transcriptional AND gate in the chloroplast of Chlamydomonas reinhardtii |
| 13:50 | Co-lab OpenPlant - interdisciplinary workshops of science art and design |
| 14:00 | Desktop plant experiment box |
| 14:10 | Environmental sensor networks based on plant electrical signalling. |
| 14:20 | Coffee Break |
| 14:50 | Plant electro-mechanics |
| 15:00 | Advancing the ability to image single RNA molecules at the cellular level |
| 15:10 | Establish a Procedure for Rapid Identification of Genetic Parts for Use in Algal Biotechnology |
| 15:20 | Establishing 3D Printed Microfluidics for Molecular Biology Workflows |
| 15:30 | Universal precise large area colony scanning stage with measurement and selection tool integration |
| 15:40 | Development of an Open Source Autonomous Imaging Station for Distribution in High Schools, Universities, and Emerging DIY Scientific Communities. |
| 15:50 | Printable SynBioLab - a feasability study |
| 16:00 | A synthetic biology approach to investigating arbuscular mycorrhizal symbiosis in Marchantia paleacea |
| 16:10 | Closing remarks |
Open Source Hardware in Synthetic Biology
Source: Open Source Hardware from the PLOS SynBio Community, licensed under CC-BY 4.0
AddThis Sharing Buttons above
by Tobias Wenzel, who received an OpenPlant Fund grant to develop DokuBricks
Open Source Hardware is an exciting new trend

Figure 1: OpenFlexture Microscope by Richard Bowman, UK, 3D printed and operated with a Raspberry Pi computer.
“This microscope is one of the cleverest pieces of open source hardware for laboratory use that I have seen so far” said Prof. Jim Haseloff in the Plant Science Department at the University of Cambridge, as he learned about a design by Richard Bowman. Richard, a fellow in the Department of Physics, was frustrated about the difficulty of working with conventional technologies for some of his experiments. For example fitting a growth chamber into a microscope can be a major challenge. Many conventional scientific instruments are designed to be incompatible to possible extensions and are not co-developed by the user community, leading to impractical designs. Thus he built his own digital microscope from a 3D printed chassis and a simple Raspberry Pi computer. The main challenge to solve for high-quality microscopy results was to position and stabilise the sample. He solved this with a one-piece 3D printed flexing mechanism in plastic. Now the microscope is ready for all sorts of adaption, it can even be placed inside commercial incubators or used in education and field uses – all at a tiny fraction of the price of conventional digital microscopes. What a great demonstration of the uses that are enabled by rapid prototyping methods! Increasingly available methods are e.g. 3D printing and laser cutting along with accessible electronic units such as Arduino microcontrollers or Raspberry Pi computers.

Figure 2: OpenTrons pipetting robot for open source liquid handling, designed by junior MIT scientists, USA
Richard is not alone in his desire to make experiments more reproducible, and customisable as well as automated. In the last few years, Open Ephys and Backyard Brains started providing tools for electrophysiology measurements, OpenTrons and open syringepump designs target the automation of liquid handling, the electrochemical potentiostat Cheapstat was a creation of an iGEM team and was published in 2011, a different iGEM team in 2015 created a fluorescence microscope adaptation of Richard’s initial design to join the ranks of another popular fluorescence microscope by Tom Baden and Andre Chagas from the University of Tuebingen, Germany. The last mentioned scientists also curate the PLOS Open Source Hardware collection where many other examples can be discovered.
Sharing equipment plans is an opportunity for experimental science, especially Synthetic Biology
Another noteworthy collection that demonstrates the fast raising popularity of Open Source Hardware is Joshua Pearce’s Appropedia and his book Open-source Lab. He illustrates the benefits of open scientific information sharing to the reader in depth. Beyond the arguments of openness, Open Source Hardware works well in science since many experimental set-ups are prototypes that are suitable for manual or rapid manufacturing methods and because technical training is widely available in science. Furthermore, it is a job requirement to demonstrate impact in this profession. Extending the publication-like sharing philosophy to hardware has worked well for the pioneers.
Synthetic Biology specifically can benefit from the open community engineering approach to bring the users and designers of complex interdisciplinary equipment closer together. The exchange of designs and protocols comes hand in hand with an increase of reproducibility of experiments, which is a major challenge of the field. The reproducibility is further enhanced by the additional number of eyes that can spot errors in protocols and improve processes without additional development cost. Open Source Hardware works well in businesses too, when equipment is expensive and specialised as is the case for most synthetic biology instruments. For example: OpenIOlabs is a young Cambridge company that (among others) is in the process of open sourcing many expensive equipment parts from the supply chain, in order to make their key products more accessible and IO Rodeo offers open source laboratory equipment for which the user can decide how many parts he wants to buy and what to build on their own. Users then often contribute improvements for free, which has most prominently driven the development cycles of 3D printers.
Many designs come from the DIY community. Are the bio laboratory designs good enough?

Figure 3: NinjaPRC, a thermocycler design for open source DNA amplification, by Shingo Hisakawa, Japan
A lot of existing open source designs come from the do-it-yourself (DIY) and maker community, as the trend only recently reached academia and business more widely. A group of scientists and makers in Cambridge, UK, tried to replicate open source designs for biology laboratory equipment to kickstart a DIY biolab and evaluate the designs along the criteria of safety, quality, adaptability and ease of build. Funded by a mini-grant of the Synthetic Biology Strategic Research Initiative, they attempted designs for electrophoretic gel boxes, a centrifuge, PCR thermocyclers, tube holders, syringe pumps and a 3D printer (more to be found online, soon). Unfortunately, the team discovered more problems than solutions: even in the documentation sets that looked good, essential parts were missing. Some designs referred to specific parts that are not generally available and that had to be shipped from the US for high costs. Most design files where difficult to adapt to other sizes of e.g. acrylic sheets when build from a metric rather than US-imperial material stock. Assembly sets that could be ordered commercially were complete and had decent to good assembly instructions, but the documentations were usually not sufficient to build the hardware without buying the set, thus not fully deserving the label ‘Open Source Hardware’.
Open Source Hardware + good documentation; it just got easier with DocuBricks
There is a lesson to be learned from the incompleteness of commercial assembly-set documentations: Open Source Hardware is more than an assembly instruction. It is also about documenting design files and decisions along its functionality and in a modular fashion, complete with testing and calibration instructions. A good documentation enables the project to grow and improve without the doing of the inventor. Only in this way most projects can enfold their benefit well to society and technology companies. To be sure, documenting a hardware project is not easy and requires time. For this reason a handful scientists at the University of Cambridge (including the author), all with a background in technology and biology, recently started the DocuBricks initiative. DocuBricks is an open source and free software that makes documenting hardware and usage procedures easier. The name is a reference to modularity in the same way as Lego or BioBricks. As the name suggests, the editor part of the software guides the user through a modular documentation structure with relevant fields in a standardised, yet general format. The user can create a hierarchy of documentation bricks, explaining their function, implementation and assembly while referring to a parts library. The result is a XML document and a folder with construction and media files that is displayed with the viewer part of the software (a style sheet and script to enable interactivity).

Figure 4: DocuBricks* logo, the new open source tool for easy generation of high-quality hardware and procedure documentations. (*Image rights: Tobias Wenzel)
Over the database with the same name, DocuBrick.com, the projects can be shared and found. The team puts emphasis on impact tracking and acknowledgement, to make engagement in Open Source Hardware not only easier, but also more promising to the scientific community. Projects can also be cited via DOI’s, which are internationally curated short links and the standard way of referencing scientific literature. The new initiative is growing fast: Richard Bowman’s microscope from the beginning of this article can already be found. The wireless communication designs of Shuttleworth Fellow and collaborator Luca Mustafa will be released soon, just like several projects of the OpenPlant Initiative in Cambridge and a number of other scientific interest groups. Even a journal on Open Source Hardware from the Ubiquity Press will be launched and intends to use DocuBricks as the preferred documentation format. When will you join the effort to make science more open and reproducible?
Tobias Wenzel is a PhD candidate at the University of Cambridge and DocuBricks founder. This blog is a summary of a presentation given at Cafe Synthetique in November 2015.
LinkedIn | Web page | ResearchGate
AddThis Settings Begin AddThis Sharing Buttons below